Research
Past departmental cohorts, scholar groups, and collaborators.
How an Ancient Process could Explain the Present
Near the end of the Pliocene, the Sierra Nevada mountains in California underwent some dramatic changes. Tectonic action that was already well underway began accelerating in earnest, raising the mountains to new heights. By the time the Pleistocene began around two million years ago, the peaks loomed imposingly over the San Joaquin Valley. Glaciers began to advance and recede in approximately 41,000-year cycles, isolating animals and plants into refugia. (More recently, the periodicity of this changed to 100,000-year cycles, when the eccentricity of earth’s orbit shifted.) The biogeographic consequences of these glacial cycles were huge: for example, over 73% of amphibian and squamate species that transect the Sierra Nevada exhibit lineage divergence in this ecoregion.
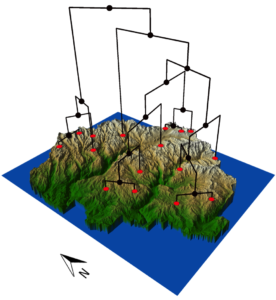
Species endemic to alpine environments often evolve via steep ecological selection gradients between lowland and upland environments. However, alpine environments have faced repeated glacial episodes over the past 2 million years, further fracturing these endemics into isolated populations. I introduced a “glacial pulse” model of alpine speciation, in which cycles of allopatry and ecologically divergent glacial refugia both play roles in generating biodiversity. I tested for patterns of glacial pulse lineage diversification in the Yosemite toad (Anaxyrus canorus), an alpine endemic tied to glacially influenced meadow environments. Using double-digest RADseq across a major portion of the species range, I identified nine distinct lineages with divergence times ranging from 214–732 ka, coinciding with multiple Sierra Nevada glacial events. Three of these lineages are products of “lineage fusion”, meaning two other lineages produced them by admixture. Demographic models suggest that these fused lineages have persisted throughout past glacial cycles, and hence they are viable despite their hybrid origin.
Multiple measures of genealogy shape supported the hypothesis that some lineages recolonized Yosemite from east of the ice sheet, whereas other lineages remained in western refugia. Interestingly, I found evidence that low- and high-elevation lineages have repeatedly adapted to divergent climatic niches. Multiple pairs of lineages were isolated into low (west) and high (east) refugia, and do not appear to freely interbreed upon secondary contact.
There are two important takeaways from these results:
- Lineage divergence has involved both allopatry, and ecological divergence.
- Lineage fusion (via secondary contact zones) is an under-appreciated phenomenon. Fused lineages could be important crucibles of adaptive diversity across deep evolutionary time.
Some technical details about the genomics and bioinformatics: This project was a huge undertaking. Using double-digest RAD-sequencing, I genotyped 650+ individuals at 3000+ RAD haplotype loci. In total, 1.88 billion reads were returned from 7 lanes of an Illumina HiSeq 2500 run. This included 161.68 gigabases (Gb) of useable sequence data with a mean quality score of 35.57. That’s over 50x the size of the human genome! Traditional methods typically pick one SNP per RAD locus, and discard the remainder of SNPs. This is quite a bit of wasted information. I wrote a very extensive python script (fasta2genotype.py) that leverages haplotype information from entire sequences. The analyses involved in this project included: concatenated and species-coalescent phylogenetics, species network analyses, demographic modeling from the joint site-frequency spectrum (SFS), spatial modeling from the SFS, Bayesian skyline analyses, MaxEnt refugial reconstruction, and tests of niche divergence.
Genomic Geography: “Islands” and “Rivers” of Speciation
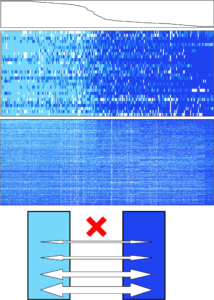
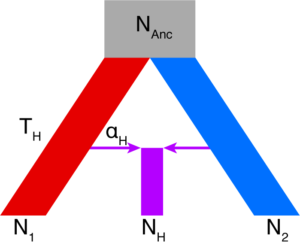
My second chapter addresses the genomic significance of the secondary contact zones identified earlier. Traditionally, and with several notable exceptions, hybridization and admixture between incipient species have been considered destructive or nuisance forces in evolutionary biology. This is because some genes may have fixed for alternate alleles, and various hybrid combinations of these may be disadvantageous. These “genomic islands” are often the exceptions to genome-wide patterns of gene flow and various levels of compatibility, and often are related to the speciation process (e.g. color pattern genes in Heliconius butterflies). In contrast to “islands”, some genes under divergent selection in the two species may actually confer a benefit to hybrids, and thus flow freely across the contact zone. These “genomic rivers” (as I am coining them) wash beneficial alleles into new genomic contexts, a process known as adaptive introgression (e.g. beak size genes in certain Darwin’s finches).
Little is known about the gene identity of genomic islands and rivers, whether there is any overlap (e.g. some genes may be neutral islands, but secondarily become adaptive rivers), or whether observed patterns are similar among different contact zones. Using the three contact zones I discovered in Yosemite National Park, and the newly minted ddRADseq dataset, I compared and contrasted the two marker types. Islands were identified with a multi-pronged set of outlier tests, and rivers were identified using Bayesian genomic cline analysis. I then constructed a de novo Yosemite toad larval transcriptome using three tadpoles from across Yosemite, to identify RAD markers that fell within gene regions, and assess whether genomic islands/rivers differed with respect to gene ontology, numbers and types of SNPs (e.g. synonymous vs. non-synonymous), and severity of protein change (e.g. missense vs. nonsense mutation).
Finally, I examined whether any of the above outlier markers related to growth and development rates of Yosemite toad tadpoles. Tadpoles in rapidly desiccating ponds are pitted with an important life history tradeoff— “get big” versus “grow fast”—choosing the correct strategy can quickly become the difference between survival and death. Yosemite toads in particular are plagued with a tendency to choose very shallow ponds, and hence desiccation and climate change are very relevant to their biology. Luckily there is phenotypic plasticity in this complex trait, allowing individual tadpoles to divest metabolic resources to match current pond conditions. We know there is a large genetic component that also governs this trait, exemplified by species such as the Couch’s spadefoot that often metamorphose in just one week! The success of these two strategies appears to be an adaptive genetic tradeoff, and several studies have suggested that genetic admixture benefits individuals specifically under scenarios of pond desiccation. Therefore I decided to examine whether genomic islands, rivers, or both were related to this critical Yosemite toad trait, by conducing a genome-wide association study (GWAS).
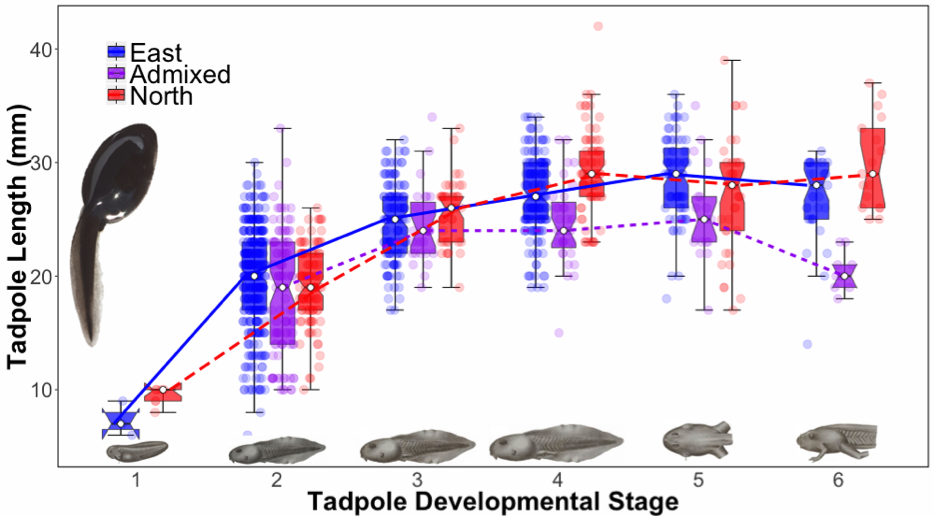
My preliminary growth and development data are consistent with the hypothesis that admixed larval Yosemite toads are better adapted to pond desiccation, and perhaps to climate change. If true, this would offer an entirely different paradigm for designing recovery plans of this and similar species that are threatened by climate change. Under the rapidly changing conditions of climate change, adaptation by mutation and selection might be insufficiently slow for some organisms to adapt and persist. However, when populations are isolated and then reunite, adaptation by introgression of novel, recombinant genotypes can catalyze adaptation. Subsequent work should use modeling, simulations, and transplant experiments to test the hypothesis that adaptive introgression is an essential evolutionary process for producing novel phenotypes in short time spans.
Some technical details about the genomics and bioinformatics: Genomic islands were identified using FST, DXY, and a cladistic metric called Slatkin and Maddison’s ‘S’. Bayesian genomic clines were estimated with bgc. Rates of asymmetrical gene flow were estimated with migrate-n for each contact zone as well. Transcriptome creation involved sequencing over 225M reads for three tadpoles on an Illumina HiSeq 2500, and a lengthy bioinformatic pipeline of assembling and annotating all isoforms with Trinity, and then calling and annotating all SNPs using GATK and Ensembl’s VEP. I discovered over 517,000 SNPs transcriptome-wide. The GWAS was performed on 1,725 individuals using generalized linear models, following best practices as defined by Reed et al. (2015).
A Novel Approach to Conservation Genomics
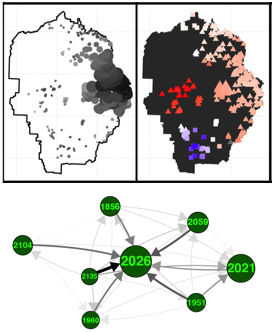
Landscape genetics is the investigation of how habitat, landscape topography, and climate influence connectivity between populations. My third chapter contributes a novel approach to landscape genetic analysis, by integrating the complex population structure among conservation units identified in the first chapter, in a genome-wide context. Genome-wide patterns of differentiation among populations of organisms are broadly created by four processes (i) limited dispersal, (ii) environmental constraints on dispersal, (iii) incompatible adaptations among populations, and (iv) founder effects. Landscape genetic studies often focus on (i) and (ii), implicitly assuming that only present-day environment is relevant, while failing to account for (iii) and (iv). Accounting for all of these factors will improve our understanding of what evolutionary processes structured natural populations over what time scales, improving future predictions of these patterns.
This approach first identifies the most likely corridors between Yosemite toad breeding meadows using Least Cost Path (LCP) analysis and causal modeling, then extracts remote sensing data from sources such as LANDSAT and the BCM climate model, and finally uses a technique called gravity modeling to identify the likely drivers of gene flow between meadows. The gravity modeling approach is particularly well-suited to pond breeding amphibians, because it models both the influence of dispersal environment and source (meadow) environment. Given the large effect that climate change is projected to have on Yosemite toads, I then forecasted graph-theoretical measures of network connectivity (overall gene flow structure) into future climate scenarios.
Delimiting Conservation Units for Evolutionary Potential
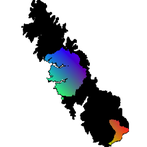
Conservation units should be drawn based on future expectations of population persistence, which depend on (i) ancient lineage boundaries representing incipient species, (ii) levels of adaptive introgression between lineages, and (iii) current levels of gene flow between populations. Therefore, all of the aforementioned work was synthesized in this chapter to predict the future of Yosemite toad persistence. I took a novel approach to conservation unit delimitation by considering putatively adaptive loci. Traditionally, conservation units are delineated based on whole-genome patterns of genetic isolation. Units that are sufficiently isolated are typically assumed to have separate evolutionary potential. However, a subset of the genome that is directly subject to positive natural selection or adaptive introgression might better reflect evolutionary potential. My method of delineating conservation units for Yosemite toads accounts for the potentially conflicting interests of preserving connectivity and promoting future adaptation to a changing environment.
I have made part of the analysis into a tutorial, which is posted here.
Field Sites
Yosemite and Kings Canyon National Parks are my primary study areas, comprising about 40% of the Yosemite toad distribution. Not a bad place for science. Approximately 99% of Yosemite toad breeding sites are meadows, either in the mid-elevation montane forest (a,c) or subalpine rocky moonscape (b,d).

Here is the Milky Way as seen from upper Kerrick Canyon in northern Yosemite, 2016. This is right before the peak of a spectacular meteor shower, the Perseids. Northern Yosemite is very remote and difficult to access for the average recreational hiker. However, starving graduate students and unpaid interns are not average…
Kings Canyon contains the very southern extent of Yosemite toads and is my other primary study site. Below you can see Goddard Canyon, where my field assistant Ross Maynard and I collected tadpole tail tips in 2011 for genetic analysis (left). We took a break to photograph two rare finds, an adult female toad (top right), and a Mount Lyell salamander (Hydromantes platycephalus, bottom right).